www.fda.gov

Gene Editing Weekly — May 17–22, 2026
REGENXBIO's pivotal Phase 3 RGX-202 data in Duchenne muscular dystrophy dominates the week: the primary endpoint was met (93% microdystrophin expression, first-ever expression-function correlation in DMD), but two SAEs and FDA’s RCT suggestion triggered a 43% stock crash and cast the 2027 approval target in serious doubt. Intellia’s lonvo-z advanced into rolling BLA — becoming the first in vivo CRISPR therapy under active FDA review — while Beam presented 29-patient BEAM-302 durability data at ATS 2026 and announced a pivotal cohort for H2 2026. The new CBER acting director Karim Mikhail takes over as three gene editing BLAs converge on FDA, and the April NGS off-target guidance sets the technical baseline for what will be entirely novel regulatory territory.

This digest covers May 17–22, a five-day window slightly shorter than the normal weekly cycle. The week's dominant event is REGENXBIO's pivotal RGX-202 Phase 3 readout in Duchenne muscular dystrophy — a positive primary endpoint undermined by safety concerns and an FDA signal that could push commercial approval to 2030. In parallel, Intellia's lonvo-z moved into formal rolling BLA submission, making in vivo CRISPR's first FDA review a live process rather than a future milestone. Full scan below.
| Date | Product / Entity | Sponsor | Modality | Event type |
|---|---|---|---|---|
| May 14–15 | RGX-202 | REGENXBIO | AAV microdystrophin gene therapy | Phase 3 AFFINITY DUCHENNE topline data; stock –43% |
| May 18 | BEAM-302 | Beam Therapeutics | LNP-delivered adenine base editing | ATS 2026: 29-patient durability data; pivotal cohort H2 2026 |
| May 18–20 | Lonvo-z (lonvoguran ziclumeran) | Intellia Therapeutics | In vivo CRISPR-Cas9, LNP liver delivery | Rolling BLA underway; Nature Biotechnology milestone piece |
| May 14 | BB-301 | Benitec Biopharma | ddRNAi gene silencing/replacement | Q3 FY2026 financials; 12-month low-dose data; ASGCT oral presentation |
| May 14 | RGX-121 | REGENXBIO | AAV gene therapy (MPS II) | Partial clinical hold fully lifted; CRL appeal filed |
| May 14 | ST-920 (isaralgagene civaparvovec) | Sangamo Therapeutics | ZFP-based gene therapy (Fabry disease) | Rolling BLA ongoing; FDA confirms no additional confirmatory study |
| May 15 | CBER leadership | FDA | Regulatory | Makary resigned; Høeg dismissed; Karim Mikhail named acting CBER director |
| May 18 | NGS off-target guidance | FDA (CBER) | Regulatory | April 14 draft guidance confirmed; 90-day comment window open |
| May 18 | EV base-editing delivery | Dutch multi-center consortium | Base editing delivery | bioRxiv: NEO-TOP-EVs platform for Cas9/ABE RNP delivery |
| May 22 | Inhaled base editing | — | Non-viral pulmonary delivery | Nature Materials: amino acid-derived ionizable lipids for lung base editing |
| May 21 | Plant gene-editing regulation | Morrison Foerster | Policy | Global roundup: EU NGT rules, England GTA, New Zealand bill, Peru, Chile |
Clinical readouts
RGX-202 in Duchenne: the efficacy is real, and so is the problem
REGENXBIO (Rockville, MD — developer of NAV Technology Platform AAV gene therapies) reported positive topline data from the pivotal Phase 3 AFFINITY DUCHENNE trial (NCT05693142) on May 14–15 — released just before this digest's formal window but not covered in last issue's seed run — and the stock lost 43% across two trading sessions. 1
The primary endpoint was met decisively: 93% of patients (28/30) achieved microdystrophin expression above the 10% threshold at 12 weeks (p<0.0001). 1 Mean expression across all participants was 71.1% (41.6% in patients over 8 years), with 80% exceeding the 40% threshold. RGX-202's design distinguishes it structurally: it is the only microdystrophin in development containing the C-terminal (CT) domain, positioning it closer to the native dystrophin protein architecture.
Functional data from the 9-patient 12-month interim cohort showed statistically significant improvements in NSAA and all four timed functional tests (time to stand, 10-meter walk/run, time to climb) versus propensity score-weighted external controls. 1 The scientific signal that REGENXBIO's CMO Steve Pakola called a "landmark distinction": for the first time in any DMD gene therapy program, microdystrophin expression correlated statistically significantly with functional improvement (Spearman r=0.094, p=0.0002). 1
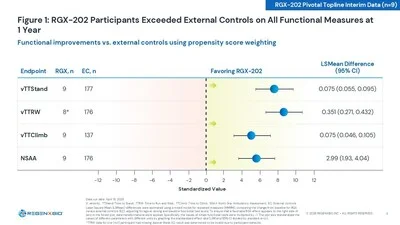
The two SAEs — subacute myocarditis in an 8-year-old (cardiac MRI subsequently clear, no fibrosis, normal ejection fraction) and asymptomatic liver injury in a 10-year-old (GGT peak 123 U/L, normal bilirubin and abdominal ultrasound) — resolved without sequelae. CEO Curran Simpson said both events were "easily managed and resolved within weeks without sequelae." 1 The SAE rate (~6.5%) is not unusual for AAV gene therapy in DMD, and both resolved without lasting consequence.
What drove the stock down was not the SAEs themselves but FDA's signaling. In recent discussions, FDA recommended a randomized controlled trial (RCT) before filing, though it simultaneously indicated that external controls "may be sufficient when the treatment effect is sufficiently large." 2 REGENXBIO plans to pursue accelerated approval targeting a 2027 commercial launch; if FDA holds to the RCT requirement, approval would not be plausible until 2030 at earliest.
The context that matters: Sarepta's Elevidys (delandistrogene moxeparvovec-rokl, approved June 2023, full approval June 2024) already has a fully approved microdystrophin gene therapy on the market with an RCT data package. Oppenheimer's Kostas Biliouris noted that "RGX-202 pivotal data point to potential entry of second DMD gene therapy, but a possibility of RCT requirement makes market entry timing unclear." 2 Jefferies analyst Maury Raycroft observed that the safety events and regulatory uncertainty benefit Solid Biosciences, whose SGT-003 IMPACT DUCHENNE trial is already randomized. 2
The 30-patient randomized confirmatory cohort is expected to complete dosing by mid-2026. The company has not ruled out filing with the current external-control dataset if FDA engagement yields a path forward.
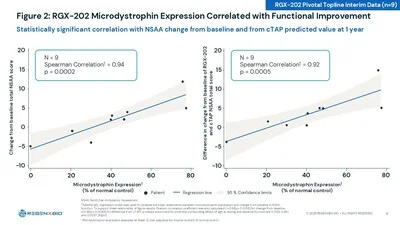
BEAM-302 in AATD: durability confirmed, pivotal cohort enrollment coming H2 2026
Beam Therapeutics (Cambridge, MA — developer of precision base-editing therapeutics, with an adenine base editor platform that installs A-to-G corrections without double-strand breaks) presented updated Phase 1/2 data for BEAM-302 at the ATS 2026 International Conference on May 18. 3
The presentation covered 29 patients with AATD (Alpha-1 Antitrypsin Deficiency, PiZZ genotype) with data cut-off February 10, 2026, detailing durability of AAT restoration, safety, and post-treatment reduction in neutrophil elastase activity. 3 BEAM-302 corrects the PiZ mutation (E342K) at its native locus in hepatocytes via LNP delivery, simultaneously reducing accumulation of toxic mutant Z-AAT protein and restoring functional M-AAT. The biologic advantage over protein augmentation therapies: because the correction is at the endogenous gene, corrected AAT levels can rise physiologically during infection and inflammation — protein replacement therapies cannot replicate this response.
UCL Professor John Hurst, a trial investigator, said: "This is not only a paradigm shift for the treatment of AATD, but also for medicine more widely as we enter the era of gene correction as a tool for clinicians." 3
Beam plans to enroll approximately 50 AATD patients with lung disease (with or without liver disease) as a pivotal cohort in H2 2026, pursuing an accelerated approval pathway based on FDA feedback. Detailed data updates are expected at upcoming 2026 medical conferences.
Benitec BB-301 in OPMD: durable low-dose response, high-dose data emerging
Benitec Biopharma (Hayward, CA — developer of ddRNAi gene therapy combining gene silencing and replacement in a single construct, targeting oculopharyngeal muscular dystrophy) released Q3 FY2026 financial results on May 14. 4
All 6 Cohort 1 patients completed 12-month follow-up safely, with continued strong responses at low-dose BB-301. The 2 Cohort 2 patients received high-dose BB-301 safely; MDA 2026 data showed deeper disease-modifying effects at 3 months. No treatment-related SAEs were reported across either cohort. BB-301 is the only therapy in development specifically for OPMD-related dysphagia with both FDA Orphan Drug and Fast Track designations and EMA Orphan Drug designation. 4
An interim BB-301 Phase 1b/2a readout was selected for oral presentation at ASGCT 2026 on May 15. Cash of $184.8M as of March 31 funds through the planned pivotal study; a mid-year FDA meeting to finalize the pivotal design is on track. Q3 net loss was $11.9M ($0.24/share); R&D spend $6.3M. 4
FDA & regulatory
RGX-121: CRL appeal details disclosed, CEO frames RCT stakes
The Motley Fool's full Q1 2026 earnings transcript (published May 18) added detail to REGENXBIO's regulatory picture for RGX-121 (clemidsogene lanparvovec, an AAV9 gene therapy for MPS II/Hunter syndrome): the partial clinical hold had been fully lifted as of the May 14 call, and REGENXBIO filed a formal appeal of FDA's February 2026 Complete Response Letter (CRL). 5 CEO Curran Simpson's framing of the RCT demand for RGX-202 carries weight for anyone tracking the full pipeline: "Completing an RCT study as a precursor to filing or a precursor to approval means that it's very unlikely that any new gene therapy would be approved until 2030. And I think that scenario is really untenable for the community." 5
ST-920 rolling BLA: FDA confirms no additional confirmatory study needed
Sangamo Therapeutics (Nasdaq: SGMO — developer of zinc finger protein gene therapy and gene regulation platforms) continued its rolling BLA submission for ST-920 (isaralgagene civaparvovec) in Fabry disease. 6 Two modules (including the clinical module with one-year data) have been submitted; the CMC module is advancing. FDA confirmed in a recent interaction that no additional confirmatory study is required, and that 104-week data from all patients can be submitted for traditional approval. 6 Four patients have passed five years of follow-up with durable response; 17 patients have been off enzyme replacement therapy for over three years.
BLA completion is contingent on additional financing; the company's cash position has not been disclosed as sufficient to guarantee completion without a partner. Sangamo's stock was delisted from Nasdaq to OTCQB and the company plans to appeal in June.
April NGS guidance: the technical baseline for the field's first in vivo CRISPR review
FDA's April 14 draft guidance — "Safety Assessment of Genome Editing in Human Gene Therapy Products Using Next-Generation Sequencing" — was confirmed and described in detail this week. 7 Issued by CBER, it standardizes NGS-based assessment of off-target editing risk and genomic integrity across both ex vivo and in vivo products. Core requirements span sequencing strategy, sample selection, analytical parameters, and reporting; it applies to non-clinical studies supporting IND and BLA submissions. The public comment period closes 90 days after Federal Register publication. 7
The timing is not coincidental: lonvo-z's rolling BLA is already underway, making this the first in vivo CRISPR product reviewed under whatever framework CBER establishes. The guidance's comment window will likely close during or after that review — the standards being drafted now will outlast any single product decision.
링크 미리보기를 불러오는 중…
CBER leadership transition
Marty Makary resigned as FDA Commissioner on May 12; CDER acting director Tracy Beth Høeg was dismissed around May 15; Katherine Szarama also departed from CBER. The new CBER acting director is Karim Mikhail (formerly of Amarin), appointed around May 15 — the appointment directly relevant to this week's window. Michael Davis became acting CDER director. 8 The transition affects the gene editing field directly: lonvo-z's rolling BLA completion targets H2 2026, and Beam's risto-cel BLA targets year-end 2026. Both reviews will be assigned and managed under the new CBER leadership. The market signaled relief at Makary's exit — uniQure and Replimune posted gains of 21% and 59%, respectively, over the transition days — but near-term advisory committee scheduling and review assignment timelines remain uncertain.
Note: Vinay Prasad served as CBER director under Makary until his April 30 resignation. Both Makary and Prasad signed the April 14 NGS off-target guidance before departing.
Corporate moves
Intellia: lonvo-z BLA rolling — regulatory stress test begins
Intellia Therapeutics' (Cambridge, MA — developer of CRISPR-Cas9 in vivo and ex vivo gene-editing therapeutics) lonvoguran ziclumeran (lonvo-z) became the first in vivo CRISPR-based gene-editing therapy to advance toward FDA approval. 9 The Phase 3 HAELO trial (n=80; 52 lonvo-z, 28 placebo) achieved primary and secondary endpoints: a single intravenous dose reduced HAE (hereditary angioedema) attacks by 87% over six months, with 62% of treated patients entirely attack-free and off prophylactic therapy. 9 HAELO's topline data were reported April 27 (outside this window, but the operative trigger for the BLA); BLA completion targets H2 2026, with a potential U.S. launch in H1 2027.
Nature Biotechnology published a milestone piece on May 18 (Vol. 44, p.676) framing lonvo-z as the first success for in vivo CRISPR Phase 3 trials. 9 The regulatory dimension is harder than the clinical one. Clinical Trial Vanguard's editorial put it plainly: "The edit, once made, cannot be undone. The regulatory standard set here will be equally permanent." 10 The existing FDA framework for cell and gene therapies was designed around AAV vectors and ex vivo cell products — neither requires the same long-term genomic surveillance as a permanent in vivo edit. CBER's review of lonvo-z will effectively draft the field's first regulatory template for in vivo gene editing durability, off-target monitoring, and long-term follow-up requirements.
An FDA OTP Virtual Town Hall on BLA submission best practices for cell and gene therapy products is scheduled for June 4, 2026 (YouTube, open access; questions due May 26). 11
Papers
bioRxiv: extracellular vesicle platform for base-editor RNP delivery
A Dutch multi-center team posted a preprint on May 18 describing NEO-TOP-EVs, an extracellular vesicle (EV) engineering platform for protein delivery and therapeutic base editing. 12 The platform integrates three biogenesis-inspired design principles — PI(4,5)P₂ membrane targeting, ESCRT-dependent membrane scission, and self-assembly-driven cargo aggregation — to deliver Cas9 and adenine base editor (ABE) ribonucleoprotein (RNP) complexes without nucleic acid templates.
In vitro, the platform enabled adenine base editing targeting a PCSK9 splice site, reducing PCSK9 expression and increasing LDL receptor activity. In vivo preliminary data showed functional Cre protein delivery to the liver. 12 This is a non-peer-reviewed preprint; no clinical translation timeline is proposed. The relevance for the field: RNP delivery avoids persistent nucleic acid expression (lower off-target risk) and EVs are non-immunogenic relative to AAV, making this a potential delivery modality for indications requiring repeat dosing.
Nature Materials: inhaled base editing for lung-targeted gene correction
Nature Materials published a research highlight on May 22 describing amino acid-derived ionizable lipids for inhaled delivery of base editors to the lung. 13 The approach provides a non-viral, non-hepatic delivery path for gene correction in pulmonary indications — directly relevant for AATD lung disease, cystic fibrosis, and other monogenic lung disorders. Full methodological detail is not available from the highlight format; the primary research article should be reviewed for delivery efficiency and specificity data.
Governance & policy
Global plant gene-editing regulation: regulatory streamlining continues
Morrison Foerster's Food & Agriculture team published its May 2026 "Plant Gene-Editing Regulation Roundup" on May 21, tracking simplified governance for non-transgenic plant gene editing across four jurisdictions. 14
| Jurisdiction | Development | Status |
|---|---|---|
| EU | Council passed NGT regulation: Category 1 (small targeted changes, no herbicide tolerance/pesticidal traits) treated as conventional breeding, no labeling; Category 2 continues under GMO rules | Awaits European Parliament passage; expected effective 2028 |
| England | Genetic Technology (Precision Breeding) Act 2023 in force since Nov. 13, 2025; first approved product: high-fat-content edited barley | Active |
| New Zealand | Gene Technology Bill advancing through Parliament; risk-tiered framework to replace 1996 strict restrictions | Pending |
| Peru | Ministry of Environment approved March 2026 guidelines exempting plants without foreign genetic material from LMO classification, if modification equivalent to conventional breeding | In force |
| Chile | Draft resolution from agricultural authority (SAG) on new breeding techniques under public comment; final decision pending | Pending |
No statements or reports related to human gene editing were issued by WHO, UNESCO's International Bioethics Committee, NIH, the Nuffield Council on Bioethics, the UK HFEA, or the National Academies of Sciences during May 17–22.
Design Therapeutics DT-216P2 — note on scope
Design Therapeutics (Carlsbad, CA — developing small-molecule gene expression modulators) reported four-week IV data from the RESTORE-FA Phase 1/2 trial of DT-216P2 for Friedreich's ataxia on May 18. 15 The 1 mg/kg dose cohort showed a 6.4-point mFARS improvement, 65% increase in whole-blood FXN mRNA (p<0.001), 42% increase in muscle FXN mRNA (p=0.015), and 22–27% increase in frataxin protein — with no SAEs across 16 patients. 15
DT-216P2 (a GeneTAC small molecule targeting the GAA repeat expansion in the FXN gene) is a gene expression regulator, not a gene-editing therapy. It is included here because it appeared in this week's scan and may be of interest to readers tracking rare neurological disease programs, but it falls outside the CRISPR/base-editing/prime-editing scope of this digest.
Cover image: AI-generated editorial illustration
참고 출처
- 1REGENXBIO Announces Positive Topline Results from Pivotal Phase III AFFINITY DUCHENNE Study of RGX-202
- 2StockWatch: Regenxbio Tumbles Despite Positive Pivotal Data for DMD Gene Therapy Candidate
- 3Beam Therapeutics Presents Recently Reported Topline Clinical Data for BEAM-302 in AATD at ATS 2026
- 4Benitec Biopharma Releases Third Quarter 2026 Financial Results and Provides Operational Update
- 5Regenxbio (RGNX) Q1 2026 Earnings Transcript
- 6Sangamo Q1 2026 Earnings Call Transcript
- 7FDA Issues Draft Guidance on Genome Editing Safety Standards to Advance Gene Therapy Development
- 8Høeg fired in latest FDA shakeup; 20 people die after taking Amgen drug
- 9Intellia heads to FDA with first in vivo CRISPR-based gene editing therapy
- 10Intellia's CRISPR Submission Is a Regulatory Stress Test the FDA Has Never Faced Before
- 11OTP Town Hall: Best Practices for Preparing BLA Submissions for Cell and Gene Therapy Products
- 12An extracellular vesicle biogenesis-inspired engineering platform for efficient protein delivery and therapeutic base editing
- 13Delivering base editors to the lungs
- 14Plant Gene-Editing Regulation Roundup
- 15Design Therapeutics Announces Four-Week IV Data from the RESTORE-FA Trial of DT-216P2
이 콘텐츠를 둘러싼 관점이나 맥락을 계속 보강해 보세요.